As part of the recent announcement of the private preview of Azure Quantum Elements, the Azure Quantum team combined new material property prediction AI models with High-performance computing calculations to digitally screen candidates for improved battery materials. By incorporating fast AI models into the screening workflow, researchers were able to expand the initial search space from thousands of material candidates to tens of millions in roughly the same time. This acceleration highlights a paradigm shift enabled by the scale and speed of Azure Quantum Elements.
Solving societal challenges requires breakthroughs in chemistry and materials sciences
More than ever, scientists need new technologies to help solve many of the most pressing issues facing society like reversing climate change, addressing food insecurity, and developing lifesaving therapeutics. Fundamentally, these problems are chemistry and materials science challenges, and some will require the transformational power of a scaled quantum computer. While we are on a path to engineer a quantum supercomputer, we are also making investments in High-performance computing (HPC) and AI to empower researchers to accelerate scientific discovery and make rapid progress toward impactful solutions for our most pressing problems today.
That is why we recently announced the private preview of Azure Quantum Elements, a comprehensive system to empower R&D teams in chemistry and materials science with scale, speed, and accuracy by integrating the latest breakthroughs in HPC, AI, and quantum computing. Researchers and product developers can screen candidates, study mechanisms, and design both molecules and materials through state-of-the-art computing capabilities and enterprise-grade services. Industry innovators, including BASF, AkzoNobel, AspenTech, Johnson Matthey, SCGC, and 1910 Genetics have already adopted Azure Quantum Elements to transform their research and development.
Scaling molecular simulations with Azure HPC
In a recent post, we highlighted how we’re scaling the applications of molecular dynamics (MD) simulations with HPC capabilities in Azure Quantum Elements. Such workloads play an important role in life sciences by simulating the structure and dynamics of proteins, the ligands bound to them, and their associated affinities. This structural exploration can accelerate the innovation of better pharmaceuticals by modeling drug molecules and their relevant protein binding sites.
In addition to applications in life sciences, MD simulations also play valuable roles in materials discovery by explaining relationships between material composition, structure, and dynamic properties. MD-calculated properties, such as thermal conductivity, ionic conductivity, and more, are often important filters in materials discovery pipelines. These MD-based filters can help researchers winnow a pool of materials candidates to a select few based on desired properties, which can then be tested in experimental settings.
With traditional HPC-based computational material discovery, density functional theory (DFT) is typically used as the engine for computing forces in MD simulations. DFT-based calculation workflows have allowed researchers to explore and evaluate thousands of materials candidates. However, these calculations come at a significant computational cost. A single static DFT calculation, for instance, can require several minutes of CPU time. Geometric optimization can demand tens to hundreds of such calculations, while MD simulations can require millions or more.
Combining HPC with AI acceleration for materials discovery
To accelerate computational materials discovery processes, we combined HPC calculations with three new AI models relating material structure to energy, force, and stress; electronic band gap; as well as bulk and shear moduli mechanical properties. The models were trained on millions of materials simulation data points to bypass HPC calculations by quickly predicting materials properties. Those capabilities allow researchers to filter material candidates based on properties like stability, reactivity, ionic conductivity, and more. When used as a force field, the AI materials models provide a 1,500-fold speedup over DFT calculations for geometric optimization of small systems with less than 100 atoms1. This speedup will be even greater for larger systems, due to the linear scaling of the AI model’s execution time with system size and the much less favorable scaling of most DFT models. This result exemplifies the power of AI to perform thousands of calculations in the time required for a single HPC simulation.
To demonstrate these acceleration capabilities, we developed a pipeline of AI- and HPC-based screening calculations allowing us to analyze tens of millions of initial candidates and narrow them down to a small sample set that best suits a particular manufacturing application. By combining both AI and HPC methods, we achieved remarkable acceleration in certain computational steps.
The AI models used for this discovery process improve upon a graph neural network (GNN)-based universal interatomic potential, trained on a massive database of structural calculations performed by the Materials Project over the past decade2. That original model achieved top accuracy in a benchmark for thermodynamic materials stability predictions with the lowest overall prediction mean absolute error3, in turn emerging as a leader for AI-guided materials discovery.
Example application to rapid materials screening
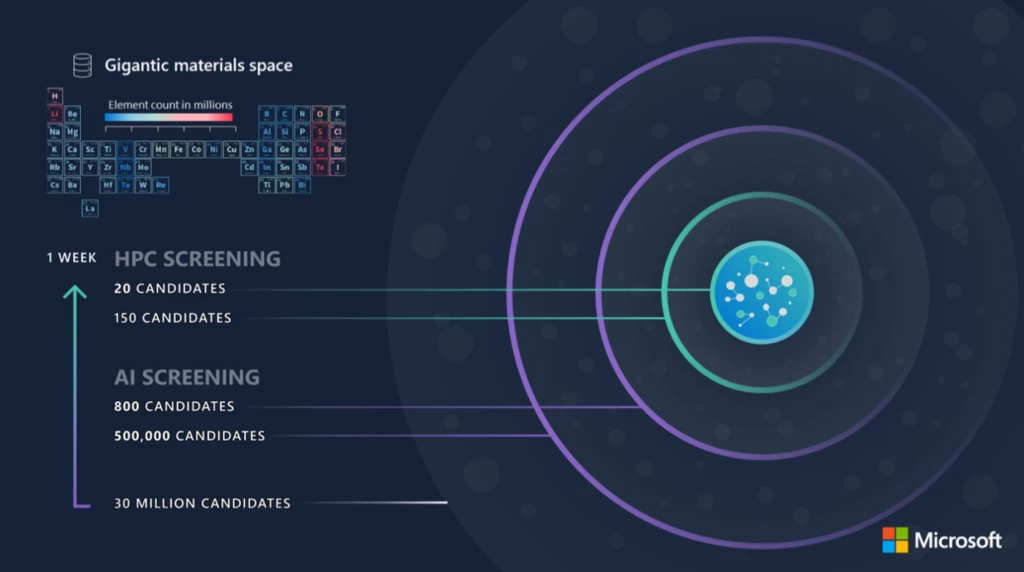
To achieve these results, we started with approximately 30 million candidate materials, generated by replacing elements in known crystal structures with a sampling of elements across a subset of the periodic table, as shown in Figure 1. We then screened this pool of candidates with a workflow that combined our AI materials models with traditional HPC-based simulations.
The first phase of screening relied on fast AI model inference calls. The AI models were used to evaluate materials stability: this step narrowed our search space from about 30 million to approximately 500,000 candidates, avoiding materials that may decompose spontaneously. The AI models were also used to screen materials for important functional properties such as redox potential and electronic band gap, reducing the search space to about 800 candidates. The second phase of screening relied on physics-based simulations accelerated with our AI models. The power of Azure HPC was used for DFT calculations to verify the properties predicted through fast AI screening in the first phase. Fast AI models have a non-zero error rate, so DFT validation re-computes the properties that the AI models predicted as a higher-accuracy filter. This verification step was followed by MD simulations to model structural fluctuations in the material. Next, we used AI-accelerated MD simulations to evaluate the dynamic properties of the materials, such as atomic diffusivity. These AI-accelerated simulations used fast AI model inference calls for forces at each MD time step, rather than the much slower traditional approach of DFT-based force calculations. This second phase of screening narrowed the field to approximately 150 candidates. From here, we assessed certain practical considerations—such as novelty, mechanical properties, and materials availability—to identify a final set of approximately 20 candidate materials worth pursuing in a lab.
This case study highlights both the scale and speed of HPC plus AI solutions as we were able to screen 30 million candidates in approximately one week, demonstrating the research acceleration that Azure Quantum Elements provides. While the work of Microsoft optimized this workflow for a specific manufacturing scenario, the materials AI models and associated HPC simulations have broad applications across diverse chemistry and materials science scenarios and demonstrates the overall feasibility of AI-accelerated materials discovery.
Azure Quantum Elements brings together years of Quantum, AI, and HPC research
At Microsoft, we see great potential to accelerate chemistry and materials advances by integrating Azure’s scaled HPC solutions with AI models tuned for scientific research. We also know that scaled quantum computing will deliver breakthrough accuracy in modeling the forces and energies of highly complex chemical systems, allowing insights into spaces that are currently intractable for classical computing. While we continue to achieve breakthrough milestones on the path to a quantum supercomputer, Azure Quantum Elements includes workflows and tools to prepare for a quantum future, providing solutions to determine which problems can be solved classically versus which require a quantum computer and estimate the number of qubits and runtimes required for various quantum chemistry calculations. Furthermore, customers can start experimenting with existing quantum hardware, and get priority access to the future quantum supercomputer from Microsoft once available.
Learn more about Azure Quantum
We are excited to see how the power of the Azure cloud will help you. For more information, please visit the following resources:
- Sign up to learn more about the private preview of Azure Quantum Elements.
- Visit the Azure Quantum Elements website.
- Read our previous blog post about Unlocking the power of Azure for Molecular Dynamics.
- Check out our Microsoft Quantum Innovator Series webinars.
1. Traditional approaches require approximately 78 CPU hours or 4,680 CPU minutes per structural relaxation. In this internal study, our AI models required a little more than 3 CPU minutes per structural relaxation, an over 1,500-fold speed up.
2. A universal graph deep learning interatomic potential for the periodic table, Nature Computational Science, 2022.
3. Matbench Discovery: Can machine learning identify stable crystals?, ICLR, 2023.