Scientists from Microsoft, ETH Zurich, and the Pacific Northwest National Laboratory have recently presented a new automated workflow to leverage the scale of Azure to transform R&D processes in quantum chemistry and materials science. By optimizing the simulation code and re-factoring it to be cloud native, the team has achieved a 30 times acceleration and 10 times cost reduction for the simulation of a catalytic chemical reaction. Moreover, these powerful automation capabilities free scientists from navigating a complex web of heterogeneous hardware and software packages, allowing them to focus on the development of new products such as sustainable production of fertilizer, more eco-friendly paints and coatings, new methods for carbon fixation, and many others.
Solving the world’s most complex and pressing challenges requires significant breakthroughs in chemical and materials sciences
Predicting chemical synthesis and catalytic processes is a key endeavor in chemistry but also poses as one of the science’s most pressing challenges. Reactions occur in a very complicated chemical space. It is almost impossible to identify the comprehensive mechanisms of chemical reactions through laboratory experiments alone. Computer simulation provides an alternative and complementary route to elucidate reaction mechanisms, but the human time involved to date has been so high that researchers have only been able to consider a few key reaction pathways that typically ignore crucial side reactions in a conventional setting. This is because the modeling of reactions requires chemical intuition and manual trial and error, and the accurate simulation of the modeled system will become intractable when the reactions are considered in full depth, including all options possible.
This reality is what motivates the Azure Quantum team every day to build a fully scalable quantum machine. Since quantum mechanics explains the nature and behavior of matter at the atomic level, quantum computers will be inherently capable of understanding and predicting the complexities of nature. While we’re making progress towards this vision, we’re simultaneously helping innovators accelerate progress in chemical and materials science today with new workflows leveraging state-of-the-art research and the power of Azure’s high-performance computing (HPC).
Introducing AutoRXN for automated reaction exploration
AutoRXN is a new automated workflow designed to empower scientists to explore reaction networks virtually using HPC in the Azure cloud. With this advancement, discovering and evaluating chemical reactions becomes extraordinarily more accessible in the cloud, which in turn will enable organizations to transform their R&D processes and speed development of new products. Using the AutoRXN workflow, scientists can expand the number of chemical reaction pathways explored from dozens to thousands of configurations with higher than conventional accuracy. The central workhorse behind the AuthoRXN orchestration is the chemical network exploration software Chemoton developed by our collaborators at ETH Zurich. We have adapted the chemistry simulations used today to be cloud-native for the modern hardware and network topology in Azure data centers, ensuring autonomy, stability, and minimum operator interference across all components of the workflow.
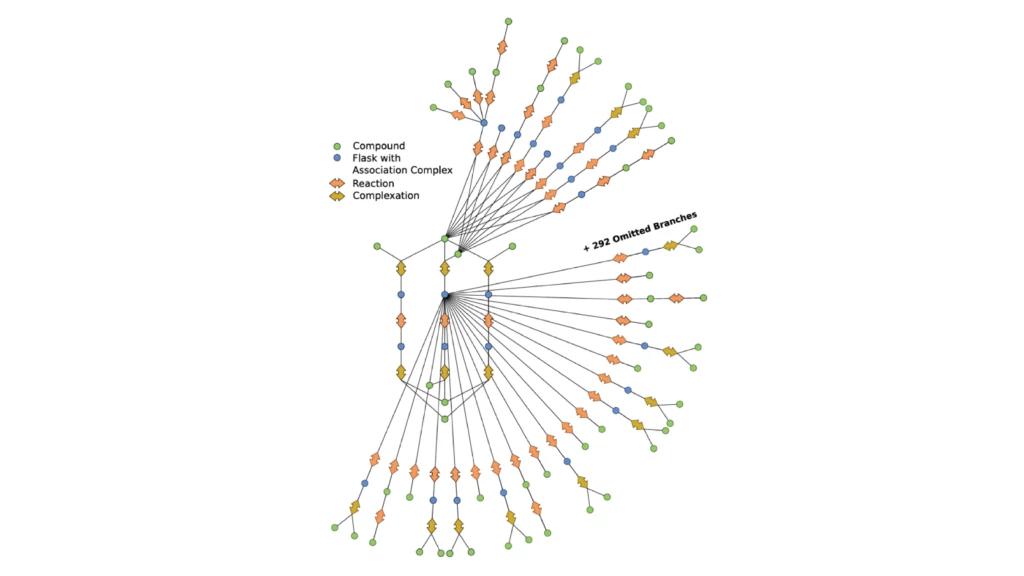
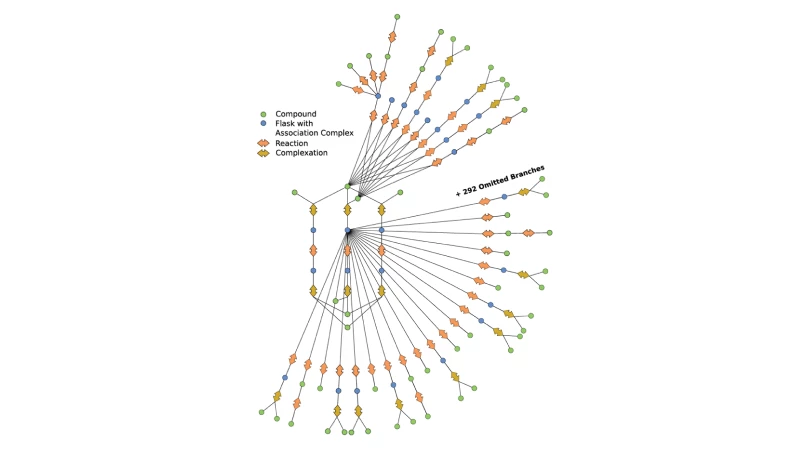
This automation enabled the research outlined in our recent paper, where we applied it to study mechanisms of an asymmetric hydrogenation catalyst. The AutoRXN workflow carries out a huge number of comparatively cheap quantum chemical calculations for exploration, automatically refines the results obtained by a vast number of expensive correlated ab initio calculations, and automates collection and evaluation of data—including back-checking of results by alternative simulation approaches. The team has been able to orchestrate highly accurate computational chemistry calculations at an unprecedented rate, which is critical for high-throughput tasks.
The AutoRXN workflow opens a new avenue of modeling and understanding chemical reactions where many side reactions can be found and studied to inform the actual performance of catalysts. The exploration scrutinizes expected reaction mechanisms and reveals the general reactivity of different atoms and functional groups in the catalyst, which enables one to improve the catalyst.
We have already identified more than five hundred reactions and more than two thousand elementary steps that reveal a comprehensive overview of the iron-complex catalyzed asymmetric hydrogenation reaction. This is far beyond the reach of conventional manual reaction modeling, as many side reactions and catalyst degradations cannot be captured by a chemist’s intuition today. Leveraging the modern hardware heterogeneity on Azure makes the process significantly faster and more cost effective. Results from the simulations help us understand the reactivity of a catalyst and accelerate R&D into exciting new discoveries in chemical and materials science.
Exploring the catalytic reactions on Azure high-performance computing provides researchers with a robust and reliable platform for hyper-scale chemistry and materials simulations without having to physically build out the system and infrastructure.
Start your path to accelerated innovation today
It’s estimated that chemistry directly touches over 96 percent of all manufactured goods.1 That means the opportunity for organizations to make new chemical and materials science discoveries to solve society’s most intractable problems and generate new growth is tremendous. New technologies and methodologies like AutoRXN are emerging from advancements in cloud computing and computational chemistry. Innovation in cloud capabilities and automation enables unprecedented scalability and hardware heterogeneity, and deep collaboration between industry and academic research is fostering the development of cloud-optimized simulation codes and methodologies. These technologies have advanced computational chemistry to a stage where it can solve the challenging problems scientists have been working on for decades.
We’re excited to see how innovators leverage the hyperscale of the cloud today and a scaled quantum machine in the future to discover new materials to solve today’s seemingly unsolvable problems and launch the next wave of technological and societal progress.
Learn more
See the publication: High-throughput ab initio reaction mechanism exploration in the cloud with automated multi-reference validation.
Explore the benefits of Azure high-performance computing in the cloud.
If you are interested in co-innovation opportunities, please contact us at QuantumInnovation@microsoft.com.
1Source : https://www.americanchemistry.com/chemistry-in-america/news-trends/press-release/2021/us-specialty-chemical-markets-start-third-quarter-on-a-strong-note