Quantum computers have the potential to solve the world’s hardest computational problems and alter the economic, industrial, academic, and societal landscape. In just hours or days, a quantum computer can solve complex problems that would otherwise take billions of years to solve.
To unlock these potential applications, the Microsoft Quantum Development Kit includes a new chemistry library that allows chemists to simulate molecular interactions and explore quantum algorithms for real-world applications in the chemistry domain. Features included in the library are state-of-the-art Q# implementations of methods for Hamiltonian simulation – including Trotterization and Qubitization techniques – state preparation techniques, and samples to help chemists to get started quickly.
For example, the 100-year-old Haber-Bosch process, a key industrial process to create artificial fertilizers, might be improved using computational chemistry methods that are enhanced by quantum computing. That could improve the catalytic process for creating ammonia from atmospheric nitrogen, a process that currently requires high temperatures, high pressures, and carefully selected catalysts. This process is so heat- and pressure-intensive that it consumes upwards of two percent of the world’s natural energy sources. Using quantum algorithms such as those supported by the Microsoft Quantum chemistry library, scientists can learn from nature’s natural process of nitrogen fixation how to achieve this task at lower pressures and temperatures.
Microsoft Quantum & Pacific Northwest Laboratory collaboration
The Microsoft Quantum Development Kit chemistry library was developed in collaboration with Pacific Northwest National Laboratory (PNNL), a leader in both chemistry and data analytics. The chemistry library – working with PNNL’s NWChem, an open-source, high-performance computational chemistry tool developed by the U.S. Department of Energy’s Office of Science – enables quantum solutions to solve computationally complex chemistry problems.
“Quantum computing has the potential to help us answer our questions much, much faster with much higher accuracy.” Wendy Shaw, Director, Physical Sciences Division, Pacific Northwest National Laboratory
Tackling hard computational chemistry problems using the quantum chemistry library
To get started in quantum chemistry simulations, the chemistry library offers features such as:
- State-of-the-art Hamiltonian simulation methods: Two main methods are supported, Trotterization and Qubitization.
- Trotterization uses the fact that fast alternation of time-slices converges to the given Hamiltonian. This typically leads to quantum circuits that implement the time evolution with a low number of qubits by cycling through all terms that are present in the given Hamiltonian in a sequential way and in a prescribed order.
- Qubitization is a technique that allows to perform an estimation of energies by way of implementing a linear combination of operators. This typically has a slightly higher cost in terms of qubits compared to Trotterization but can lead to substantially lower circuit size and circuit depth, depending on the chemistry problem at hand. A crucial part of Qubitization quantum algorithms are state-preparation techniques, which are provided as part of this release.
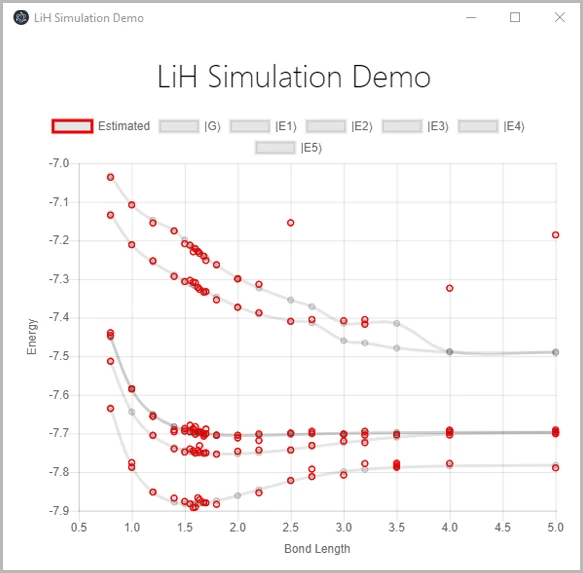
- Estimation of ground and excited state energies. The chemistry library enables the estimates of ground state and excited state energies as a function of bond distance. Many real-world chemistry problems involve not only estimates of ground state energies but also the understanding of the dynamics of the various transitions between excited states and a characterization of their energy levels. This type of problems can be studied with the new chemistry library as well.
As an example, the figure below shows the results of one of the samples that uses the data from NWChem. It graphically shows the equilibrium bonding distance of Lithium Hydride for various distances and energy levels.
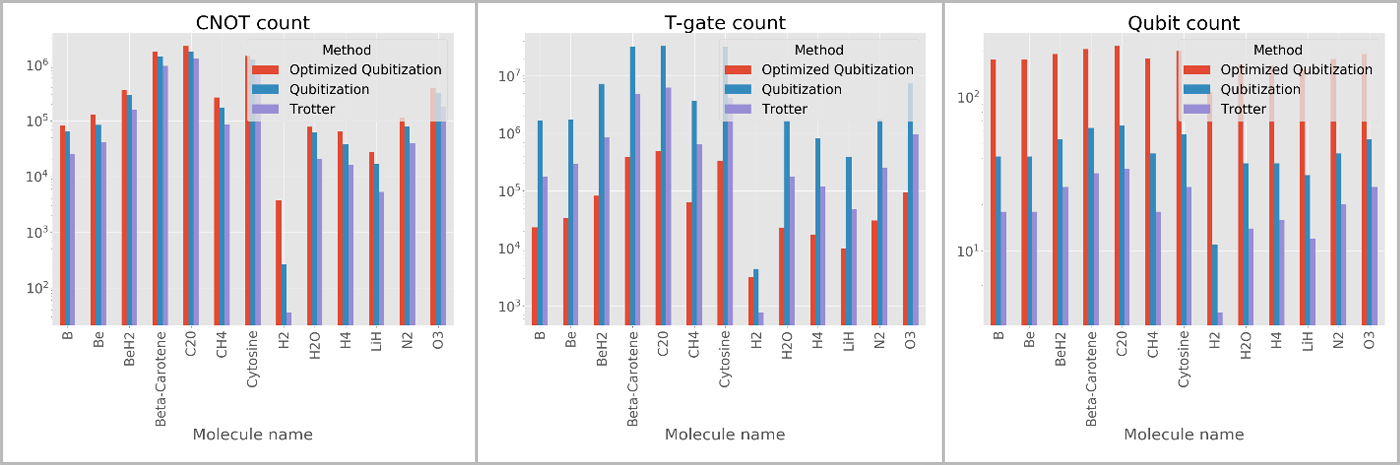
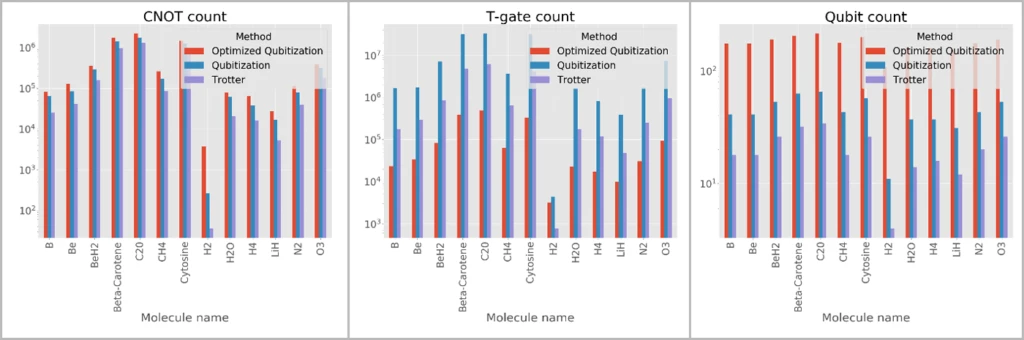
- Automatic resource estimation. The Trace simulator that is part of the Quantum Development Kit allows chemists to estimate important metrics about quantum algorithms, such as the number of qubits, the total gate count for various types of primitive gates, and the total circuit depth. Shown below is the output of the GetGateCount sample that is shipped with the library and which computes a variety of useful metrics for various molecular benchmarks that are provided as part of the library.
- Interface to powerful chemistry modeling tools. The release introduces an open source YAML-based schema called Broombridge (in reference to a landmark celebrated as a birthplace of Hamiltonians), to make it easy for a chemist to input real-world chemistry models into Q#. Broombridge is a structured, extensible, and human-readable and human-editable way of representing electronic structure problems. Learn more about the schema.
- Samples: The chemistry library contains several benchmark samples molecules using the to help programmers and chemist simulate molecules and chemical interactions. Some examples:
- Beta-Carotine: this is a benchmark to study the most common form of carotene in plants and oxidative damage; an ideal candidate for singlet fission processes.
- C20: this benchmark studies C20 systems often used to calibrate various electron correlation effects.
- Ozone: this benchmark to study quantum mechanical excited-state studies of the Ozone and an understanding its role in Earth atmosphere.
- Real-world chemistry modeling in NWChem. The chemistry library interfaces with NWChem, a high-performance computational chemistry software package. There are several ways to explore the interplay between NWChem and the Q# quantum chemistry library, including:
- Try out the sample molecules listed above that have been generated by NWChem and are already a part of the Quantum Development Kit Samples.
- Use EMSL Arrows Builder for the Microsoft Quantum Development Kit – a web-based frontend to NWChem – to generate new Broombridge-formatted molecular input files
- Use the Docker image, provided by PNNL to run NWChem and generate your own molecule models
You can learn more about the chemistry library by exploring the documentation, including a set of conceptual documentation to describe basic principles of quantum chemistry, its mapping to a quantum computer, and the API documentation.
Today, developers and chemists around the world can begin exploring the Microsoft Quantum Development Kit and experiencing the world of quantum computing. Resources such as the chemistry library will enable problem-solvers in the computational chemistry domain to explore the world of quantum. We’re excited to help our growing community take another step toward the new world of quantum computing.